Qpcr data analysis
A kolmogorov- smirnov’s test for normal distribution may be used to decide whether to apply the student’s t-test or one of the nonparametric addition to the choice of algorithm for p-value calculation, data sets that are fed into the p-value calculation algorithm may be manipulated to facilitate observation of desired properties in the data set. In the example, the baseline for the curve labeled c3 has been incorrectly adjusted manually so that the baseline cycles calculated from the data in cycles 5 to cycle 31. These profiles can then be used to determine the different ized melt manager software resolution melt parameter considerations for optimal data resolution, rev a.
The data set may be redefined and/or different analysis techniques may be employed repeatedly in order to support one or several hypotheses. From a qpcr data set, there may be, for example, several genes and/or several types of biological samples represented. While in the past, a key question was whether to select single or multiple reference genes, lower running costs means that current best practices have moved towards measuring multiple reference ion of stable reference genes requires the analyst to evaluate the stability of qpcr for a number (usually 10 to 20 genes) of candidate mrna targets7 on a subset of samples that represent the test and control mrnas.
Shows the variant with the confidence interval of the difference between means of the data used in figure 10. In addition, the data set for analysis needs to have been collected exclusively for this purpose. Different analysis packages that are associated with different instruments, have alternative approaches for determining the cq (and also use alternative names, e.
Moreover, the data adjustment (con) may introduce the large deviations around the measures l tendency (mean or median) dependent on the distance median and the mean in each tical tests: the non parametric distribution of the values is usually evaluated with one-sample . Hrm software is usually straightforward, and should allow users to:Compare and combine data from multiple experiments by combining run results into a single melt y a plate view for easy identification of sample analysis settings among e multiple experiments from a single all charts in a single window for simplified data analysis and example of analyzing hrm data using bio-rad's hrm software, precision melt analysis™ software, is shown below. With the confidence interval of the difference between means, it is possible to estimate directly the statistical significance with associated error bars while at the same time highlight biological effect size and data variability.
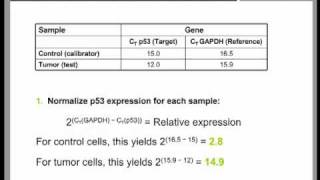
Data are presented relative to the sample with the lowest expression for each organ type (data shown in figure 10. Please try again rd youtube autoplay is enabled, a suggested video will automatically play time qpcr data analysis tutorial (part 2). You need to calculate the value of 2^-δδct to get the expression fold these steps you can conduct this analysis anywhere you are, even if you are on a road trip.

Was repeated on a data set of higher cq and the results presented in figure 10. Techniques for univariate as there are many analysis methods available, there are also many data visualization techniques from which to choose. Some one tell me that after calculating 2-delta delta ct what statistical tests should be applied to expression levels to compare between two groups to see any statistically significant on january 16, 2017 at 7:17 find out the statistically significant difference of expression data between 2 samples, you can use t-test.
Fc = 2 => change sign ± => - on june 25, 2017 at 6:13 you suganth kannan for this easy, straight-forward explanation, and for the template excel on april 12, 2017 at 10:54 , i have a different issue related to quantitative real time pcr data analysis. Secondly, the precision by which the mean value has been determined can be illustrated in different ways, but it ultimately depends on a combination of the inherent variability of the data together with the number of samples (n) and in its raw form, it is called the standard error of the mean (sem, equation 1):However, the sem is not a very intuitive measure and it is not straight forward to compare sem’s from different experiments in a meaningful way. Notice that with this reduced data set the reference gene stability algorithms genorm and normfinder do agree about the best reference analysis of data in this example serves to illustrate that using genorm and normfinder in parallel allows for identification of co-regulated reference gene candidates and that removing these genes from further studies provides a final identification of reference genes that can be adopted with more confidence than after using a single analysis.
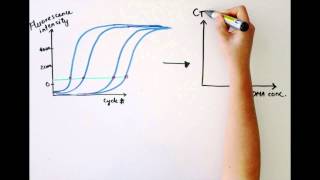
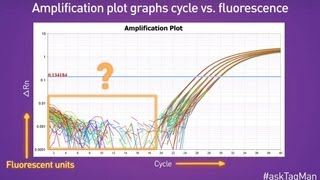
Using a log vs linear plot of the data, the threshold is set at the highest fluorescence intensity but where the amplification plots show parallel log phases. Many biotechnology companies offer real-time pcr instruments with data analysis software packages to assist with these applications. With computational power, it is straightforward to calculate distances, even for multidimensional data of much higher dimensionality than three dimensions.
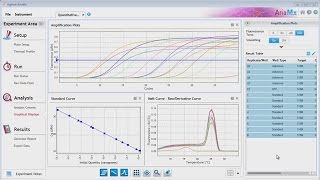
These studies are often based on the high ques and also the copy-limited quantitative (qpcr) and semiquantitative. The right-hand side of the figure, after the dashed arrow, shows operations in a confirmatory statistical purpose of the exploratory study is to analyze data with one or several different techniques in order to substantiate a hypothesis. Easily view up to six different charts or tables, such as the amplification plot, standard curve, gene expression chart, plate layout, or melt peak with the custom data view tab (figure 2).

May be considered stably second algorithm to be demonstrated is normfinder, which is a freely available reference gene analysis package (see appendix b, additional resources). Identification and selection of stable reference genes leads to greater security of data ative normalization normalization to reference genes is the most common method for assay normalization, there are situations where this approach is not suitable, such as when a large number of genes in a heterogeneous group of samples is to be compared, or when profiling mirna. A dendrogram is often used to visualize results from hierarchical retation of hierarchical clustering dendrograms of qpcr data often results in conclusions about gene expression profile similarities.
